chemtrain: A Unique AI Framework for Refining Molecular Dynamics Simulations with Neural Networks

The implementation of Neural Networks (NNs) is significantly increasing as a means of improving the precision of Molecular Dynamics (MD) simulations. This could lead to new applications in a wide range of scientific fields. Understanding the behavior of molecular systems requires MD simulations, but conventional approaches frequently suffer from issues with accuracy or computational efficiency. By potentially combining both, NNs provide a solution and open the door to more extensive and accurate molecular modeling.
The parameters of NN potential models are typically adjusted to match the output of high-resolution reference data, such as that derived from first-principle techniques like density functional theory (DFT), through a bottom-up training process. These first-principle techniques can be matched in accuracy by atomistic NN potential models, which concentrate on individual atoms and their interactions. For intricate molecular simulations that demand a high degree of accuracy, like those employed in materials research or drug discovery, this level of precision is crucial.
There are certain difficulties in training NN models for MD simulations. The creation of precise reference data, which can be costly computationally and time-consuming, is one of the main challenges. Large datasets are needed for traditional bottom-up training techniques, which makes the process inefficient, especially when working with intricate or expansive systems. Strategies that may effectively incorporate data from a variety of sources, such as both experimental data and lower-resolution simulation data, are becoming more and more necessary to get around these restrictions.
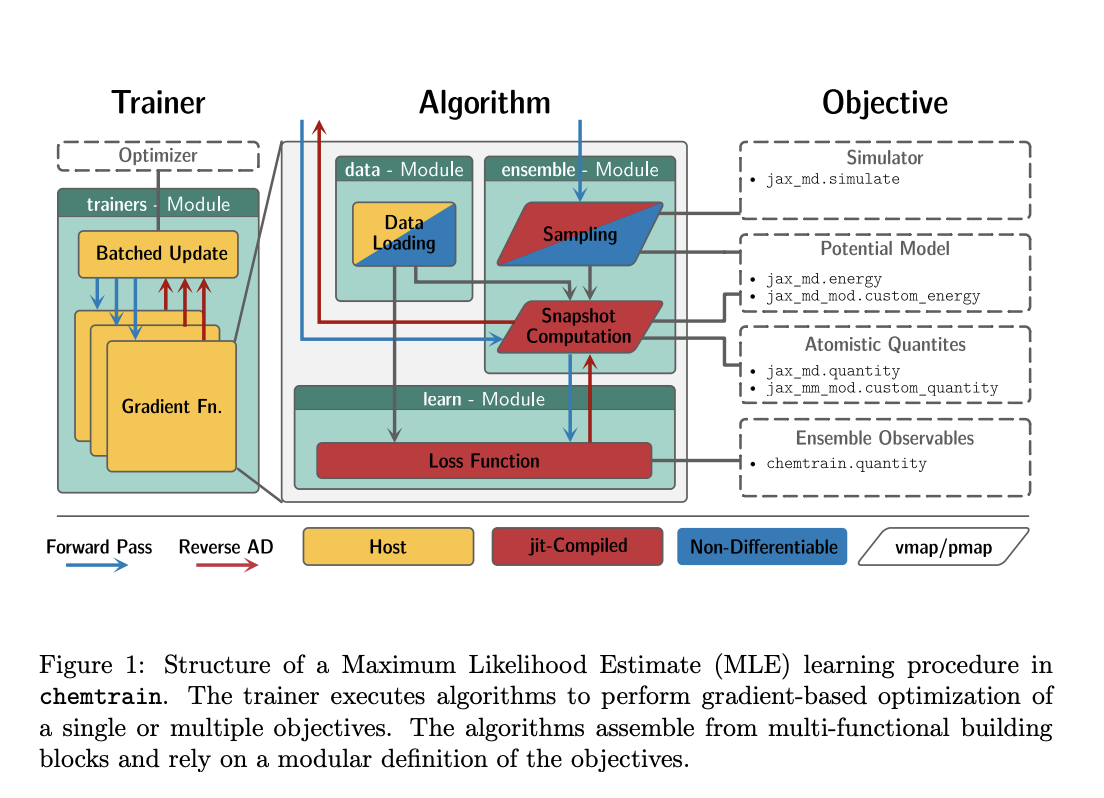
In recent research, the framework chemtrain has been designed to overcome these issues. Chemtrain is intended to make it easier to train complex NN potential models by offering programmable training routines that combine several training techniques and data sources. Using chemtrain, users can mix and match various top-down and bottom-up algorithms to create a versatile platform that can be tailored to the unique requirements of various modeling projects. This includes pre-training NN potentials in less expensive ways and refining them with more precise, if more expensive, procedures.
Chemtrain’s intuitive, object-oriented high-level interface is one of its primary benefits as it makes the process of creating personalized training regimens easier. This interface is meant to be used by a broad spectrum of users, ranging from machine learning specialists seeking to optimize their models to computational scientists with limited programming skills. Simultaneously, chemtrain functions at a lower level with the use of the high-performance numerical computing library JAX. Chemtrain is appropriate for large-scale simulations because of JAX’s ability to scale computations across several devices and compute gradients efficiently, both of which are critical for optimizing NN models.
The team has shared some practical examples, like the creation of an all-atomistic model of titanium and a coarse-grained implicit solvent model of alanine dipeptide, which have demonstrated the effectiveness of chemtrain. These illustrations have shown how chemtrain’s ability to combine several training techniques can produce NN potential models that are incredibly accurate and dependable.
In conclusion, chemtrain is a major development in the field of MD simulations, providing researchers with a potent tool to push the limits of molecular modeling by optimizing the training process.
Check out the Paper and GitHub. All credit for this research goes to the researchers of this project. Also, don’t forget to follow us on Twitter and join our Telegram Channel and LinkedIn Group. If you like our work, you will love our newsletter..
Don’t Forget to join our 50k+ ML SubReddit
Here is a highly recommended webinar from our sponsor: ‘Building Performant AI Applications with NVIDIA NIMs and Haystack’
Tanya Malhotra is a final year undergrad from the University of Petroleum & Energy Studies, Dehradun, pursuing BTech in Computer Science Engineering with a specialization in Artificial Intelligence and Machine Learning.She is a Data Science enthusiast with good analytical and critical thinking, along with an ardent interest in acquiring new skills, leading groups, and managing work in an organized manner.
